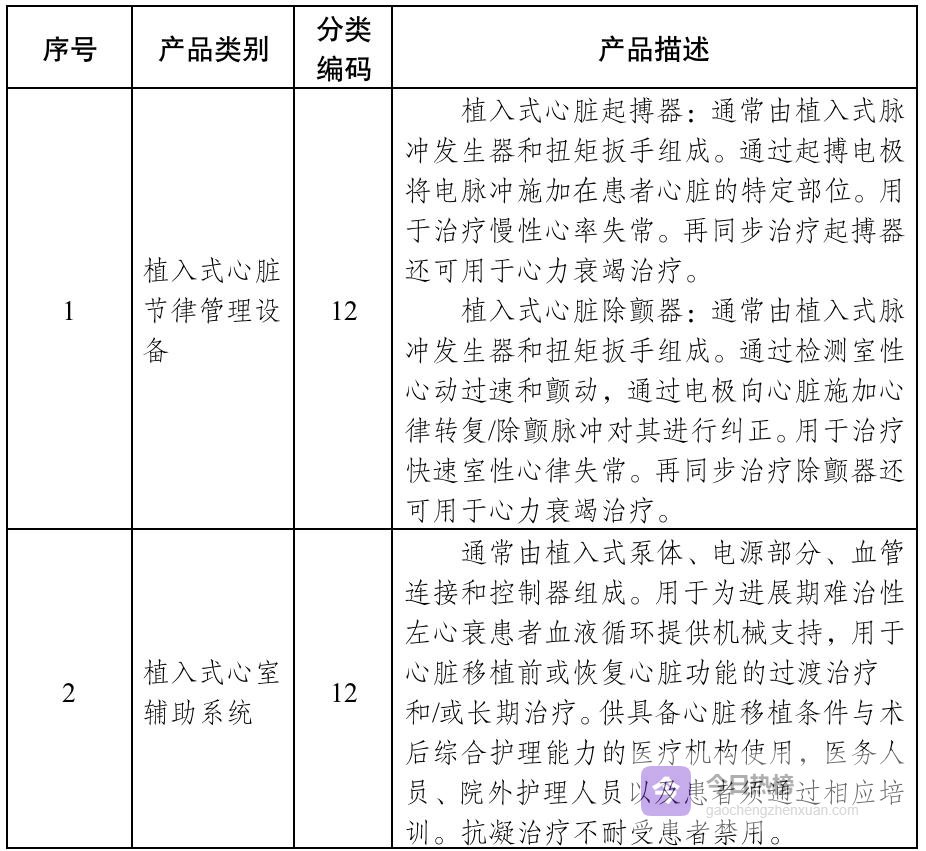
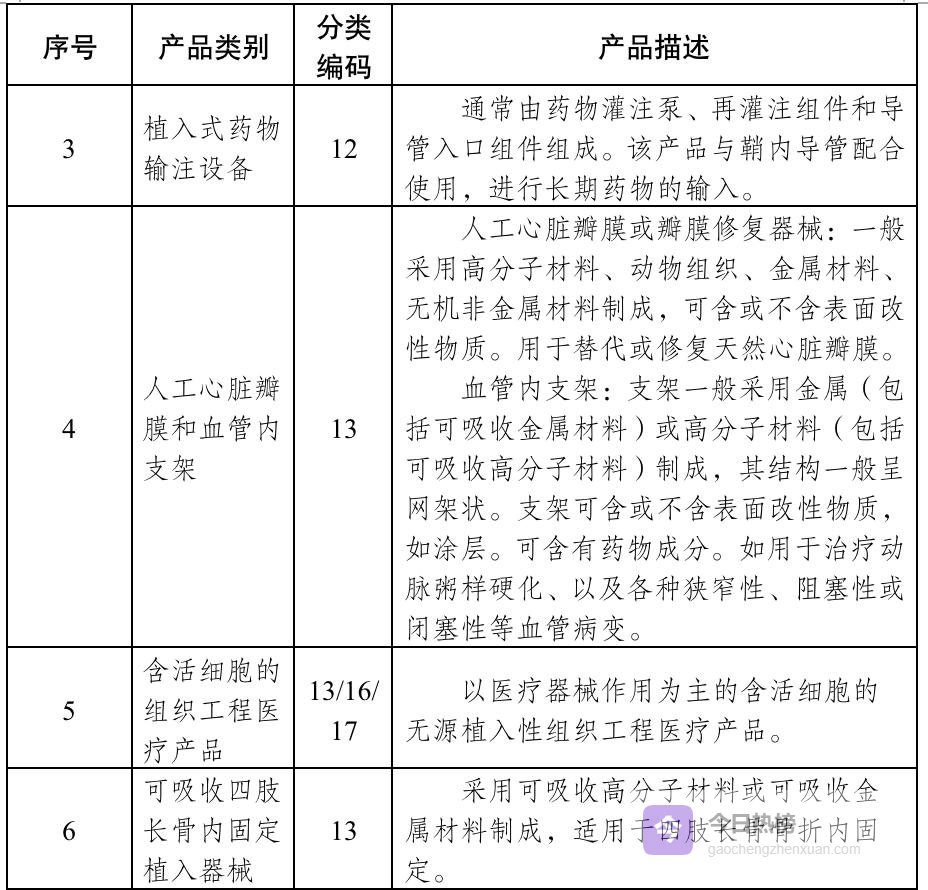
01政策速递:药监局更新高风险器械临床试验“黑名单”
9月14日,国家药监局正式发布《需进行临床试验审批的第三类医疗器械目录(2020年修订版)》,并同步废止2014年旧版通告。新规把“全新设计、材料或机理+全新适用范围+高风险”三类条件捆绑成硬性门槛,凡符合任一情形的三类器械,在中国开展人体试验前,必须先拿到国家药监局的“准生证”。
02政策背景:审评放管服与创新激励双轮驱动
本次修订的核心任务是落实“放管服”与鼓励创新”一揽子文件精神:既要给真正创新的产品开绿灯,又要把受试者安全与数据真实性牢牢攥在监管手里。通过提前锁定高风险品种,药监局希望把临床试验的“前门”开得更大,把“后门”堵得更死,既提速也提质。
03目录亮点:一张图看懂哪些产品必须“报批”
新目录并未列出成百上千个品种,而是用“原则+类别”的抽象方式给行业划出清晰红线:
全新设计、材料或机理;
全新适用范围;
人体风险等级高。
只要同时满足以上任一组合,哪怕只是微创新,也要走审批通道。目录同时明确,“已上市产品+新增适应症”不再属于强制报批范畴,为企业节省大量时间和资源。
04企业应对:早布局、早卡位、早受益
对创新器械申请人而言,“报批”不是拦路虎,而是过滤器:能证明临床价值、风险可控且方案科学的产品,将更快拿到入场券;反之,则被迫回到实验室继续迭代。因此,提前锁定伦理审查、建立受试者数据库、准备多中心协同方案,成为抢跑2021年的关键动作。
05监管逻辑:把“高风险”锁进制度的笼子
药监局在通告里反复强调“推进监管科学研究成果转化”,实质就是把人工智能、大数据、真实世界证据等工具嵌入临床试验设计与质量把控。未来,审批速度将取决于企业能否提供高质量、可追溯、可审计的临床数据;而那些仅靠“概念”或“故事”的产品,则可能永远停留在PPT阶段。
原创文章,作者:马超,如若转载,请注明出处:http://m.gaochengzhenxuan.com/news/17972.html