01封面故事:乳腺干细胞“熄火”,肿瘤自然减速

图1. Cell Reports 封面图像
正常乳腺干细胞移植后,能完整再生乳腺导管树(绿色);当这些干细胞敲除CDK14后,再生优势瞬间丧失,仅零星出现红色导管。这幅封面图直观展示了CDK14对乳腺干细胞活性的“点火”作用,也预示了抑制这条通路可能成为遏制肿瘤发生的策略。
02研究速览:CDK14成为三阴性乳腺癌“刹车片”
2.1 ❒ 文章亮点首次把非典型CDK14拉到三阴性乳腺癌的聚光灯下
在病人来源的类器官(PDO)与异种移植模型(PDX)中,敲低CDK14或用小分子抑制剂处理均可显著抑制肿瘤生长
机制上,CDK14通过强化Wnt信号通路,维持乳腺干细胞及癌干细胞的数量与功能
2.2 ❒ 实验细节研究人员先在体外把CDK14从乳腺干细胞里“抠”掉,发现细胞自我更新与成瘤潜能直线下降;再把这群“缺火”的细胞打入小鼠体内,肿瘤出现时间推迟、体积缩小,竞争性再生优势几乎归零。
进一步利用CRISPR敲除PDX模型中的CDK14,结果同样“不给面子”——肿瘤消退曲线明显优于对照组。这些数据共同证明:CDK14是三阴性乳腺癌进展的驱动因子。
03同门兄弟CDK16:走的是另一条“转移高速”
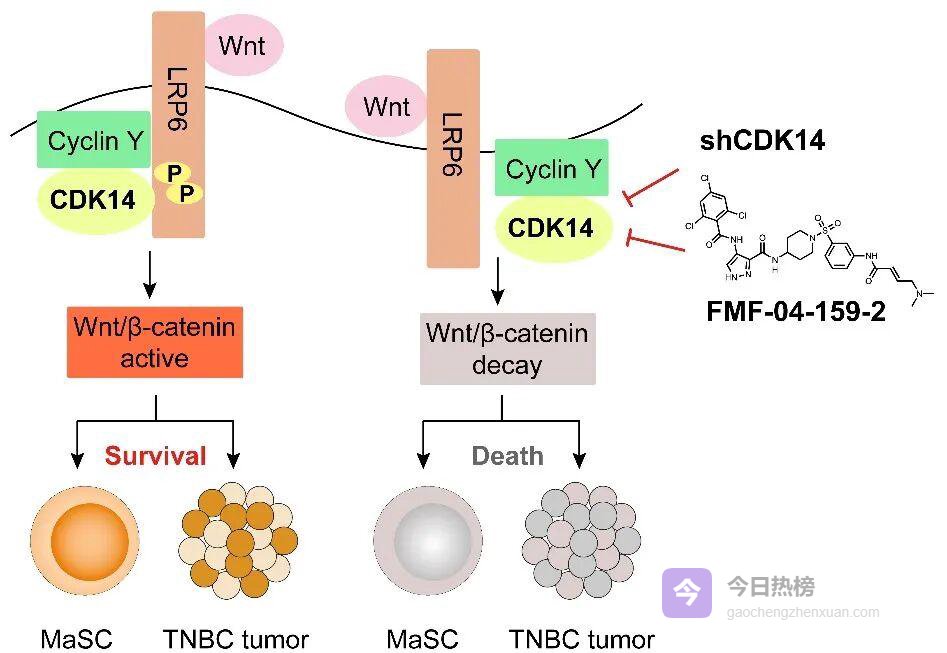
图2. CDK16调控三阴性乳腺癌转移的分子机制
当大家把目光集中在CDK14时,课题组同步扫描了另一位非典型CDK——CDK16。令人意外的是,CDK16并不走Wnt老路,而是直接磷酸化PRC1蛋白,松绑染色质压缩、激活上皮—间质转化(EMT),最终把三阴性乳腺癌推向肺与肝转移。
在裸鼠实验里,同时敲低CDK16与PRC1,肺转移结节数量下降超过60%;给已转移小鼠喂食CDK16抑制剂,转移灶回归肉眼可见。两条独立通路、两种死亡模式,为后续联合用药提供了“双保险”。
04从实验室到病房:非典型CDKs的“翻译”潜力
目前FDA已批准CDK4/6抑制剂用于HR+/Her2-晚期乳腺癌,转录型CDKs也步入临床一期。相比之下,非典型CDKs仍是一片蓝海。武汉大学团队用PDO/PDX大模型完成“最后一公里”验证:
CDK14抑制→肿瘤生长减速
CDK16抑制→转移风险下降
这为将来设计“双靶点”或“可切换”抑制剂奠定关键证据链,有望克服单一靶点耐药、延缓疾病进展、延长患者生存。
05结语:非典型CDKs的“第二幕”已拉开
从默默无闻到登上Cell Reports封面,CDK14与CDK16用短短数年完成“逆袭”。它们不走传统周期通路,却精准卡位干细胞维持与转移关键节点,为三阴性乳腺癌提供了全新的干预思路。随着抑制剂走向临床,乳腺癌的治疗菜单或许将新增一道“非典型”却高效的选项。
原创文章,作者:林诗雨,如若转载,请注明出处:http://m.gaochengzhenxuan.com/yule/9496.html


